| 服務聯繫 |
110 年 4 月 6 日公告之「醫療器材標籤應刊載 單一識別碼規定」,規定第二等級及第三等級醫療器材 之單一包裝或器材本體上,應標示單一識別碼(Unique Device Identifier, UDI)
- (一)自中華民國110年6月1日起製造之國產及輸入第 三等級植入式醫療器材。
- (二)自中華民國111年6月1日起製造之國產及輸入第三 等級非植入式醫療器材。
- (三)自中華民國 112 年 6 月 1 日起製造之國產及輸入第 二等級醫療器材。
 衛生福利部於 110 年 4 月 28 日公告「應建立與保存來源及流向 資料之醫療器材」規定,應建立與保存醫療器材來源流向資料之 醫療器材共計有 202 項醫療器材品項。 「應申報來源及流向資料之醫療器材品項」依醫療器材分類分級 管理辦法所定如下:
衛生福利部於 110 年 4 月 28 日公告「應建立與保存來源及流向 資料之醫療器材」規定,應建立與保存醫療器材來源流向資料之 醫療器材共計有 202 項醫療器材品項。 「應申報來源及流向資料之醫療器材品項」依醫療器材分類分級 管理辦法所定如下:
- (一) E.3610 植入式心律器之脈搏產生器。
- (二) I.3540 矽膠充填之乳房彌補物。
- (三) L.5980 經陰道骨盆腔器官脫垂治療用手術網片。
- 根據「醫療器材來源流向資料建立及管理辦法」第 3 條規定:
非持有醫療器材許可證或完成登錄之醫療器材販賣業者 應以電子或書面方式,依本法(醫療器材管理法)第 19 條第 1 項 規定,建立及保存醫療器材下列供應來源及流向之資料:
一、 供應來源資料:
(一) 供應者之名稱、地址及聯絡資訊。
(二) 產品識別資訊。
(三) 批號或序號。
(四) 數量。
(五) 收貨日期。
(六) 製造日期及有效期間,或保存期限。
(七) 其他中央主管機關指定之項目。二、 流向資料:
(一) 供應對象之名稱、地址及聯絡資訊。
(二) 產品識別資訊。
(三) 批號或序號。
(四) 數量。
(五) 交貨日期。
(六) 製造日期及有效期間,或保存期限。
(七) 其他中央主管機關指定之項目。
![]() 醫療器材商利用通訊交易(html)通路販售醫療器材
醫療器材商利用通訊交易(html)通路販售醫療器材
- 申請作業流程請參考國產醫療器材QMS稽核作業流程圖
- 依據:醫療器材品質管理系統檢查及製造許可核發辦法(內網)pdf(doc)
- 硬體設施符合醫療器材製造業者設置標準(內網)要求
- 準備系統文件:醫療器材品質管理系統準則(內網)
- 在醫療器材品質管理申請平台提出申請(參考:醫療器材品質管理系統檢查及製造許可核發辦法)(內網)pdf(doc)
- 應填具申請書,檢附附表一所定文件、資料,並繳納費用後,向中央主管機關提出。
- 醫療器材品質管理申請平台/
- QSD申請製造廠基本資料(英文版) (內網.doc)
- QSD申請書(英文版)(內網.doc)
- 國外製造廠實地檢查併品質系統文件審查減免須知(內網pdf)
- 有關輸入醫療器材製造廠申請品質管理系統檢查,應依醫療器材品質管理系統檢查及製造許可核發辦法第2條檢附附表一所定文件,其得減免之文件,請參照「美國廠簡化模式」、「歐盟技術合作方案」、「日本廠簡化模式」公告內容。
- 依據醫療器材品質管理系統準則第七十八條,製造業者僅生產附表所列醫療器材品項者,應就其生產之每一類型或系列分別建檔,並實施紀錄管制、申訴處理及矯正與預防措施。前項業者,除第十一條至第十三條、第四十七條、第五十五條、第六十三條、第六十四條、第六十九條、第七十六條及第七十七條規定外,不適用該準則第二章至第六章之規定。
- 有關輸入醫療器材國外製造廠之實地檢查相關事宜,請參照「國外醫療器材製造廠實地查廠申請」網頁內容。
- 符合醫療器材品質管理系統準則(QMS)之品質系統文件審查(QSD)申請書(輸入醫療器材製造業者)及製造廠基本資料(英文版)僅供國外製造廠準備文件時參考,送件時請以中文版申請書為準。
- 醫療器材製造業者設置標準(內網
- MMRDir/Annex
- 歐盟醫療器材上市要求的歐盟醫療器材法規(MDR)自2021年5月26日起正式實施,國內驗證模式亦同步修正
全世界皆然,驗證均為各國政府掌管醫器部門控管,服務申請費用
若相關資料提供不全,無法提供報價
- 歐盟醫療器材上市要求的歐盟醫療器材法規(MDR)自2021年5月26日起正式實施,國內驗證模式亦同步修正
- 應以登錄方式取得上市許可之醫療器材品項(自110年10月1日起)
- 衛生福利部註冊與線上申報平台(申報說明)
- 第一等級醫療器材查驗登記申請(附表一_申請製造-輸入第一等級醫療器材查驗登記應檢具之文件-資料.pdf)
- 第一等級醫療器材查驗登記申請文件檢送簡表(2021-04-30)(內網doc)
- 第一等級醫療器材查驗登記申請書(2021-04-30).(內網doc)
- 申請製造第一等級醫用口罩聲明書(2021-05-03)
- 第二/三等級醫療器材查驗登記申請
- 第一等級醫療器材查驗登記申請(
- 製造許可證檢附文件:(專案申請核准)
- 一、醫療器材製造許可影本(認可登錄函影本)。
- 二、醫療器材商許可執照影本。
- 審查費為新台幣15,000元整,請於申請時繳納(送案時請先繳納審查費即可)。
- .證照費為新台幣1,500元整,請於領證時繳納。
- 一、醫療器材製造許可影本(認可登錄函影本)。
- 免取得醫療器材製造許可品項.pdf
- 醫療器材委託製造申請及相關事項(受託者應先取得醫療器材製造許可後,委託者始得申請醫療器材委託製造)
- 醫療器材品質管理系統檢查及製造許可核發辦法
- (一) 本項文件指由中央主管機關核發之醫療器材製造業者符合醫療器材品質管理系統準則之證明文件影本。
- (二) 原列屬藥品管理者,於公告改列醫療器材之日起三年內,得以符合藥品優良製造規範之證明文件影本替代之。
- 國內醫療器材製造業者品質管理系統準則條款QMS
- 醫療器材標籤、說明書或包裝擬稿
- 醫療器材商執照
- (一) 製造醫療器材者,應檢附醫療器材製造業許可執照影本。輸入醫療器材者,應檢附營業項目包含“輸入”之醫療器材販賣業許可執照影本。
- (二) 國內委託製造者,應檢附委託者及受託製造業者之醫療器材商許可執照。
- 出產國許可製售證明(國產者免附)
- 國外原廠授權登記書(國產者免附)
- 臨床前測試及原廠品質管制之檢驗規格與方法、原始檢驗紀錄及檢驗成績書
執行檢測生物相容性、電性安全性、電磁相容性檢測及無菌性試驗之受託實驗室,應符合下列條件之一:- 符合 ISO/IEC 17025 之規定。
- 符合藥物非臨床試驗優良操作規範(GLP)之規定。
- 產品之結構、材料、規格、性能、用途、圖樣等有關資料完整技術性資料
- 產品之結構、材料、規格、性能、用途、圖樣等有關資料:儀器類產品者,得以涵蓋本項資料之操作手冊及維修手冊替代之。
- 醫療器材安全性與功效性基本規範及技術文件摘要/【發布日期:2021-04-28】
- 醫療器材安全性與功效性基本規範及技術文件摘要(Essential Principles and Summary of Technical Documentation, EP/STED).doc
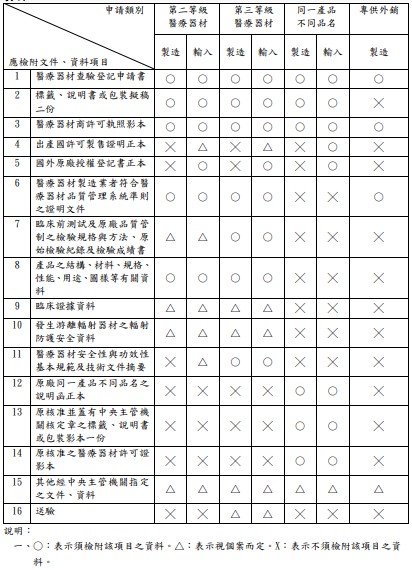
- 第2、3等級查驗登記申請書
- 醫療器材標籤、說明書或包裝擬稿
- 醫療器材商執照(專案申請核准)
- (一) 製造醫療器材者,應檢附醫療器材製造業許可執照影本。輸入醫療器材者,應檢附營業項目包含“輸入”之醫療器材販賣業許可執照影本。
- (二) 國內委託製造者,應檢附委託者及受託製造業者之醫療器材商許可執照。
- 出產國許可製售證明(國產者免附)
- 國外原廠授權登記書(國產者免附)
- 臨床前測試及原廠品質管制之檢驗規格與方法、原始檢驗紀錄及檢驗成績書
執行檢測生物相容性、電性安全性、電磁相容性檢測及無菌性試驗之受託實驗室,應符合下列條件之一:- 符合 ISO/IEC 17025 之規定。
- 符合藥物非臨床試驗優良操作規範(GLP)之規定。
- 產品之結構、材料、規格、性能、用途、圖樣等有關資料完整技術性資料
- 產品之結構、材料、規格、性能、用途、圖樣等有關資料:儀器類產品者,得以涵蓋本項資料之操作手冊及維修手冊替代之。
- 醫療器材製造業者符合醫療器材品質管理系統準則之證明文件
- (一) 本項文件指由中央主管機關核發之醫療器材製造業者符合醫療器材品質管理系統準則之證明文件影本。
- (二) 原列屬藥品管理者,於公告改列醫療器材之日起三年內,得以符合藥品優良製造規範之證明文件影本替代之。
- 第2、3等級輸入(國產)醫療器材查驗登記提會案件專用資料表
- 醫療器材安全性與功效性基本規範及技術文件摘要/【發布日期:2021-04-28】查檢表
- 醫療器材安全性與功效性基本規範及技術文件摘要查檢表(Essential Principles and Summary of Technical Documentation, EP/STED).doc/內網pdf
- 醫療器材人因可用性工程評估指引.pdf
- FDA Human Factor and Medical Device /
- IEC 62366-1 AMD 1-2020.xdf/Medical devices - Part 1: Application of usability
engineering to medical devices
- IEC TR 62366-2-2016.xdf/Medical devices - Part 2: Guidance on the application of usability engineering to medical devices
- IEC 60601-1-6 AMD 2-2020.xdfMedical electrical equipment – Part 1-6: General requirements for basic safety and essential performance – Collateral standard: Usability
- (come from IMDRF/pdf, International Medical Device Regulators Forum)
- ISO 14971:2019 Medical devices — Application of risk management to medical devices
- 政府公告
- 療器材優良臨床試驗管理辦法
- 軟體確效IEC 62304 Software life cycle
- 政府公告
- 醫療器材軟體確效指引.pdf
- FDA Guidance for the Content of Premarket submission
for Software Contained in Medical Device
- 適用於製造廠之醫療器材網路安全指引
- 電性安全試驗
(Electrical Safety test)
IEC 60601-1:2005/ AMD1:2012/ISH1:2021 .pdfMedical electrical equipment –
Part 1: General requirements for basic safety and essential performance
- 電磁相容性試驗
(Electromagnetic
compatibility test)
IEC 60601-1-2:2014/ AMD1:2020 CSV.pdfMedical electrical equipment –
Part 1-2: General requirements for basic safety and essential performance –
Collateral Standard: Electromagnetic disturbances – Requirements and tests
- 生物相容性試驗 (Biocompatibility )
ISO 10993-1(2018)
ISO 10993-5(2009)
ISO 10993-10(2010)
- 功能性試驗 (Performance test)
- 醫療器材人因可用性工程評估指引.pdf
- 各類產品
- 認證實驗室查詢
- 疫苗進口:根據指揮中心公布的指引,第一步要先委託藥商辦理相關程序,要提出8項資料檢送,包含執
- 行計畫書、
- 數量及計算依據、
- 供貨期程、
- 有效期限、
- 藥品說明書、
- 冷鏈及倉儲設備、
- 原廠授權書、
- 外國上市證明或替代文件。
- 醫療器材安全性與功效性基本規範及技術文件摘要查檢表(Essential Principles and Summary of Technical Documentation, EP/STED).doc/內網pdf